About
Molpher-lib is a C++/Python library based on the program Molpher, which implements systematic chemical space exploration using a method called molecular morphing (original publication available here). However, the purpose of Molpher-lib is not just molecular morphing implementation, but it also strives to become an easy-to-use de novo drug design tool whose capabilities go beyond the original Molpher and offer wider spectrum of applications.
On this humble web page, we will shortly explain what molecular morphing is, how it can be used in de novo drug design and we will hint at some advantages Molpher-lib has over Molpher. If you want to skip this part and dive directly into the code examples, there is a page dedicated to it, which should help you quickly understand how the library works and what it can be applied for. Of course, there is also the documentation, which includes a more extensive tutorial.
Molecular Morphing
Molecular morphing is an atom-based de novo drug design method that uses stochastic optimization to search for a 'path' in chemical space. This path is simply a set of molecular structures that result from iterative application of small structural modifications to an input molecular structure. An example of such a path can be seen in the following image:

Example of a chemical space path generated by Molpher from an input structure by iterative application of modifications (so called 'chemical operators'). The operators used in this example are shown as codes below the arrows in this figure (RA = remove atom, RB = remove bond, BR = bond reroute, MA = mutate atom).
The original Molpher software was written for the task of finding a set of modifications needed to transform one molecular structure to another. Therefore, it can generate a set of 'hybrid' structures that combine structural features of two input molecules; very much like morphing in computer graphics.
One possible application of an algorithm like this is sampling unknown chemical space 'around' molecules with confirmed biological activity (see figure below). Here, Molpher (or Molpher-lib) can be used to generate a focused virtual library of hybrid structures that are derived from known active molecules. Since these hybrid structures combine features of bioactive molecules and, thus, occupy similar area of chemical space, it is very likely that the hybrid molecules will likely have activity of their own. Therefore, virutal screening against this focused library of hybrids could be more successful tha against a random library.
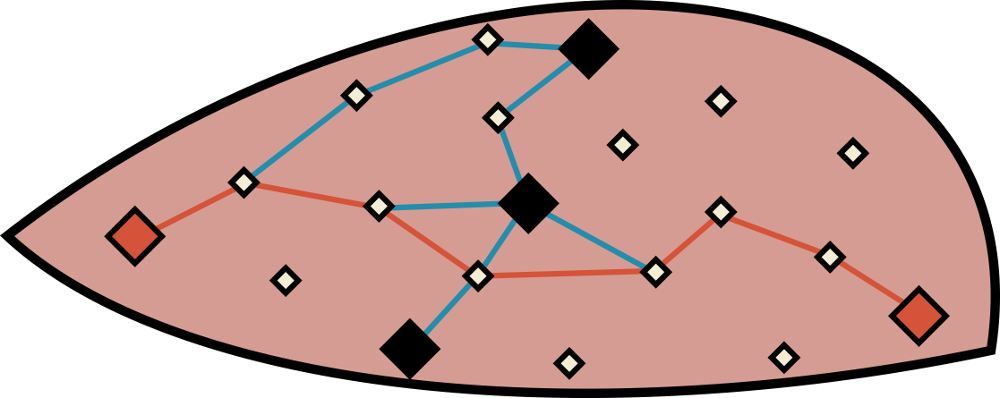
Schematic depiction of a focused chemical space exploration experiment using molecular morphing. Multiple paths are generated between every pair of known active molecules. The lines in this picture correspond to a morphing operator while diamonds represent molecular structures. The big diamonds are then known active molecules between which we are trying to navigate. The small diamonds represent hybrid structures that are generated during morphing. A chemical space path example is highlighted in red. Because we are staying 'close' to the original input structures, generated hybrids are more likely to fall into the 'active' subset of chemical space (represented by the thick black lines enclosing the red area).
Molpher-lib vs. Molpher
The motivation to develop Molpher-lib was mainly driven by various shortcomings of Molpher:
- Molpher was developed as a standalone user-oriented software with no easy programmatic access to its features.
- Molpher only includes one exploration algorithm implementation with very limited possibilities for customization.
- Molpher is written purely in C++ which makes experimenting with new exploration strategies difficult and interfacing with software in other languages more difficult.
- The software architecture is not modular enough which makes extending the C++ codebase a lengthy and difficult task.
Molpher-lib attempts to address these issues as follows:
- Remove all unnecessary and outdated parts and focus on development of good API, which can be used to build more complex applications. Therefore, the GUI was removed and steps were taken to encapsulate more of the original Molpher features and expose them in the Molpher-lib API.
- User should be able to easily access and modify the chemical space exploration process. Therefore, the library defines interfaces that make it easy to inject customized code to any stage of the process (see tutorial sections on morphing operators and tree operations). This makes Molpher-lib a highly customizable framework and new exploration algorithms were already implemented with it as a proof of concept.
- The C++ interface of Molpher-lib is exposed to Python and the library comes with a convenient Python package that also offers additional functionality and new algorithm implementations. This makes the library makes interfacing with other scientific packages from the Python ecosystem easy and straightforward.
- Molpher-lib attempts to have modular software architecture that divides its functionality in small independent chunks. This makes modification, replacement and integration of new functionality much easier than before.
Furthermore, Molpher-lib includes new features that were not present in Molpher. A notable addition is the ability to lock certain atoms against certain types of modifications. This allows the user to explore chemical space around a particular scaffold or generate paths with molecules that have certain structural patterns conserved.
Documentation
At the moment, the newest non-development version of the library is v0.0.0b3. Documentation for each main version of the library and some development snapshots can be found through the following links: